Lysosomes are subcellular organelles that degrade macromolecules and pathogens, and are important for nutrient recycling and signalling. The intraluminal pH of lysosomes is low, and their content of acid hydrolyses and Ca 2+ high. Hence, rupture of lysosomes poses a potential hazard to cells. Given the detrimental consequences of impaired lysosomal integrity and the high frequency of lysosomal membrane permeabilization in diseases and normal aging, cells have evolved essential mechanisms to rapidly repair damaged lysosomes. When severely damaged, lysosomes are engulfed by autophagic membranes in the process known as lysophagy, which is initiated by the recognition of luminal glycoprotein domains by cytosolic lectins such as Galectin-3.
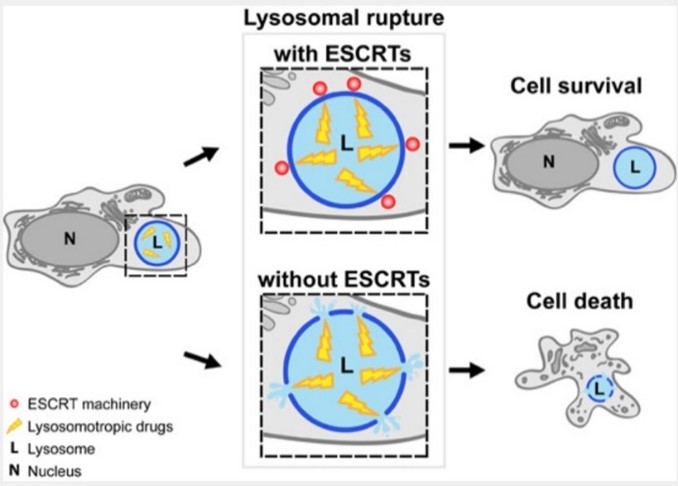
We have shown that various lysosome membrane injuring conditions trigger recruitment of the endosomal sorting complex required for transport (ESCRT). This recruitment precedes Galectin-3 and the lysophagy machinery. Interference with ESCRT recruitment impairs lysosome repair and causes otherwise reversible lysosome damage to become lethal (Figure 1).
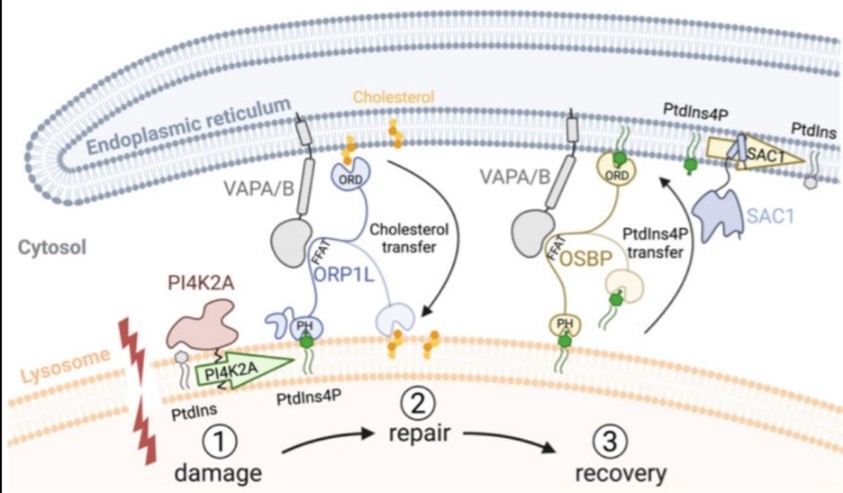
Recently, we described an additional mechanism for lysosome repair that is activated independently of ESCRT recruitment. This process involves rapid formation of contact sites between damaged lysosomes with the endoplasmic reticulum (ER) (Figure 2). Briefly, the ER contacts are activated on damaged lysosomes in parallel to ESCRTs to provide lipids for membrane repair, and that PtdIns4P generation and removal are central in this response.
We are interested to understand the mechanism of lysosome repair and how ruptured membranes are sealed. We are also investigating additional mechanisms for lysosome repair, which protect against lysosomal damage-induced cell death.