Dravet syndrom er en alvorlig form for epilepsi som oppstår i tidlig barndom. Etter en kort periode med normal utvikling får pasienter med Dravet syndrom epileptiske anfall allerede rundt et halvt års alder, ofte i forbindelse med feber (også kalt feberkramper).
I løpet av de første leveårene rammes pasientene gjerne av ulike typer epileptiske anfall som sterkt påvirker den nevrologiske utviklingen, og de blir ofte diagnostisert med ulike adferdsforstyrrelser, inkludert autistiske trekk.
I majoriteten av pasientene (70–80 %) finner man mutasjoner i SCN1A genet, som er ansvarlig for en type natriumkanaler som man finner i hjernens nerveceller. Blant flere typer ionekanaler er natriumkanalene spesielt viktige for dannelsen av raske elektriske signaler i sentralnervesystemet.
Oppdagelsen av sammenhengen mellom SCN1A-genmutasjoner og Dravet syndrom førte til klassifisering som en kanalfeilsykdom («channelopathy»).
Oppdagelsen ledet også til flere nye forskningsprosjekter som dreier seg om hvordan mutasjoner i enkelte ionekanal-gener forårsaker epilepsi og nevrologiske/psykiatriske sykdommer.
I samarbeid med våre internasjonale samarbeidspartnere forsker vi på de underliggende mekanismene bak hvordan feil i SCN1A-genet kan føre til forandret hjerneaktivitet.
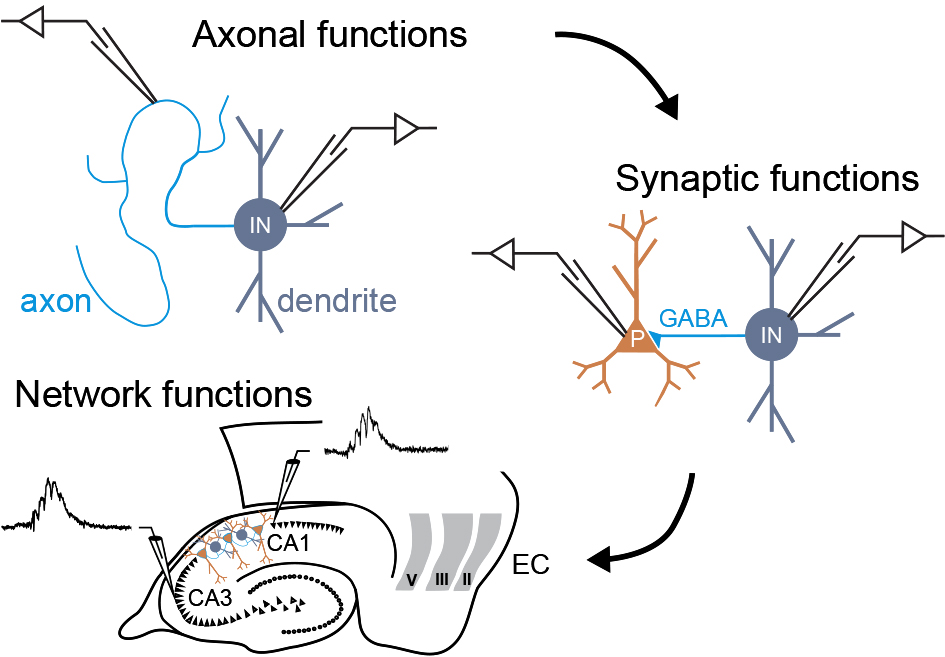
Basert på vår kunnskap om inhibitoriske (hemmende) nerveceller, som inneholder en stor andel av SCN1A-baserte natriumkanaler, skal forskningsgruppen måle elektrisk aktivitet fra natriumkanaler og kompleks aktivitet fra nettverk av nerveceller i hjernevev fra mus.
Vi bruker genetisk modifiserte mus som mangler halvparten av SCN1A genet (Scn1a+/- mus), og som utvikler lignende symptomer som pasienter med Dravet syndrom.
Å studere effekten av ionekanal-dysfunksjoner er viktig for å bidra til forbedret behandling av pasienter med disse lidelsene, og også for å forstå grunnleggende prosesser i kompleks hjerneaktivitet.